Collected the videos in a playlist
1,338 views • Nov 9, 2024
Was recently asked about HGVS nomenclature reporting. The fun thing about biology is that there's going to be exceptions to the rule or some shenanigans that you didn't expect when setting out a rule.
"The Human Genome Variation Society (HGVS) provides standardized recommendations for describing human sequence variants, which are widely accepted in the scientific community, especially in the practice of clinical molecular pathology.1 Use of the HGVS nomenclature system is a de facto recommendation for clinical reporting of sequence variants.2, 3 Being a core component of the clinical report, incorrect HGVS nomenclature can have a negative impact on patient care, such as misdiagnosis or clinical trial ineligibility. HGVS nomenclature has been traditionally computed manually by pathologists from Sanger sequencing electropherograms. However, manually computing HGVS nomenclature is time consuming, complex, and error prone, particularly with insertion and deletion (indel) variants, resulting in inconsistencies across laboratories."
Source:Clinical Implementation and Validation of Automated Human Genome Variation Society (HGVS) Nomenclature System for Next-Generation Sequencing–Based Assays for Cancer
In the 25th Anniversary Special Issue of Human Mutation, Den Dunnen et al. (2016) publish an update of the Human Genome Variation Society (HGVS) recommendations for the description of sequence variants (http://www.HGVS.org/varnomen). One of the issues discussed is how widespread HGVS nomenclature is used and, when used, whether published variant descriptions correctly follow the recommendations. An EGFR (OMIM# 131550) lung cancer testing scheme assessed in January 2016 by the United Kingdom National External Quality Assessment Scheme (UK NEQAS) for Molecular Genetics demonstrates the current variability in the use and interpretation of the HGVS guidelines by diagnostic laboratories based across the globe.
Shall explore this tool
hgvs: A Python package for manipulating sequence variants using HGVS nomenclature: 2018 Update
https://github.com/biocommons/hgvs
Reproducible, scalable, and shareable analysis pipelines with bioinformatics workflow managers | Nature Methods
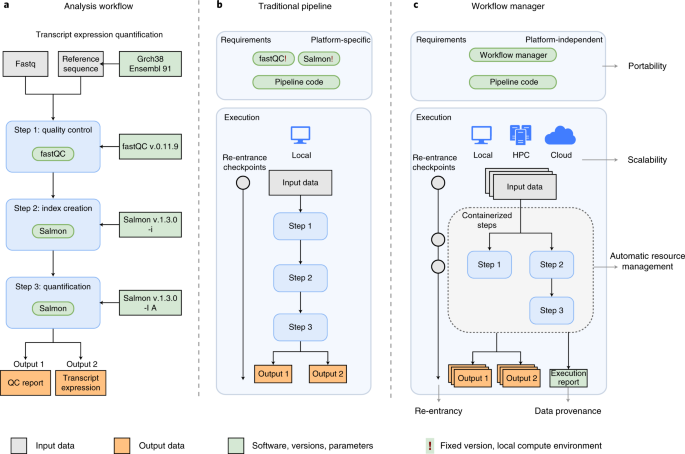 | The rapid growth of high-throughput technologies has transformed biomedical research. With the increasing amount and complexity of data, scalability and reproducibility have become essential not ... www.nature.com |
Workflow managers provide an easy and intuitive way to simplify pipeline development. Here we provide basic proof-of-concept implementations for selected workflow managers. The analysis workflow is based on a small portion of an RNA-seq pipeline, using fastqc for quality controls and salmon for ... github.com |
strand bias and orientation bias – GATK (broadinstitute.org)
"
The read orientation artifact, also known as the orientation bias artifact, arises due to a chemical change in the nucleotide during library prep that results in, for example, G base-paring with A. This kind of artifact has a clear signature (e.g. C to A SNP that occurs predominantly for the middle C in the DNA sequence CCG), and it’s singlestranded in nature. Downstream, this artifact manifests as low allele fraction SNPs whose evidence for the alt allele consists almost entirely F1R2 reads or F2R1 reads. A read pair is F1R2 (forward 1st, reverse 2nd) if the sequence of bases in Read 1 maps to the forward strand of the reference (F1), and the sequence of Read 2 to the reverse strand
of the reference (R2). F2R1 is defined similarly
if someone has read the dragonbioit used guide in illumina, it just mentioned orientation bias, ignore the strand bias.
"
Just realised that other than vcf-compare and bedtools intersect
there's other options
https://github.com/RealTimeGenomics/rtg-tools
https://github.com/Illumina/hap.py
Also there's actually new variant callers ..
Molina-Mora, J.A., Solano-Vargas, M. Set-theory based benchmarking of three different variant callers for targeted sequencing. BMC Bioinformatics 22, 20 (2021). https://doi.org/10.1186/s12859-020-03926-3
Krishnan, V., Utiramerur, S., Ng, Z. et al. Benchmarking workflows to assess performance and suitability of germline variant calling pipelines in clinical diagnostic assays. BMC Bioinformatics 22, 85 (2021). https://doi.org/10.1186/s12859-020-03934-3
. verify_variants.py
Zook, Justin M et al. “An open resource for accurately benchmarking small variant and reference calls.” Nature biotechnology vol. 37,5 (2019): 561-566. doi:10.1038/s41587-019-0074-6
Python library to parse, format, validate, normalize, and map sequence variants. `pip install hgvs`
Had to work with Ion Torrent BAMs for this but I think it's applicable to everything
Needed to run this on unmapped reads so running this first.
| #!/bin/bash | |
| samtools view -b -f 4 $1 > $1.unmapped.bam | |
| samtools index $1.unmapped.bam | |
| java -Xmx8g -jar /opt/picard/picard-tools-current/picard.jar SamToFastq I=$1.unmapped.bam F=$1.unmapped.bam.fastq | |
| /home/ionadmin/bin/bbmap/reformat.sh in=$1.unmapped.bam.fastq out=$1.unmapped.bam.fastq.fasta | |
| gzip $1.unmapped.bam.fastq.fasta |
After that the next script is fairly simple
| ~/bin/kraken2/kraken2 --db ~/k2_standard_16gb_20210517 --use-names --gzip-compressed $1 --output $1.kraken --report $1.kraken.report | |
| cut -f2,3 $1.kraken > $1.kraken.kronainput | |
| ktImportTaxonomy -tax /home/kev/Krona-master/KronaTools/taxonomy $1.kraken.kronainput -o $1.kraken.kronainput.html |